
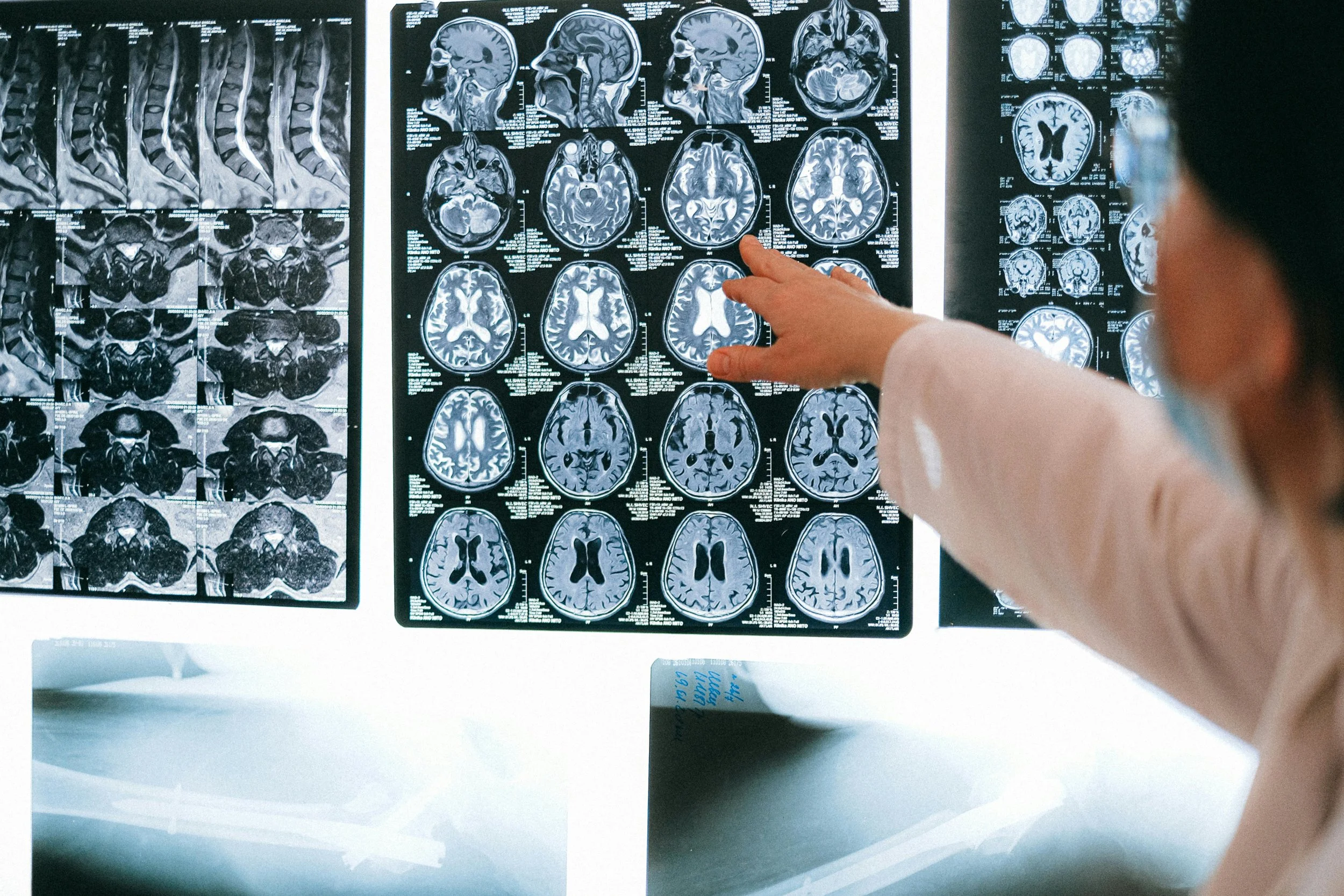
ALSP - Adult Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia
Also Known as: CSF1R Related Leukoencephalopathy; Hereditary Diffuse Leukoencephalopathy with Spheroids (HDLS); Pigmented Orthochromatic Leukodystrophy (POLD)
Overview.
Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) is a rare, progressive neurological disease that affects the brain’s white matter. White matter contains nerve cells and their fibers, which are protected by a myelin sheath and supported by glial cells.
In ALSP, the nerve fibers develop bubble-like swellings called spheroids, and the supporting glial cells become damaged. This leads to the breakdown of the protective myelin covering. As a result, ALSP can cause significant changes in personality, behavior, movement, and cognitive function.
Symptoms and progression vary widely from person to person, making diagnosis challenging. As the disease advances, it can severely impact independence and quality of life. While there is currently no cure, raising awareness and advancing research offer hope for better treatments in the future.

Causes.
ALSP is most commonly caused by a mutation in the CSF1R gene on chromosome 5. This gene provides instructions for making the CSF-1 receptor protein, which plays an important role in cell growth, division, and development.
The mutation disrupts these signaling pathways, preventing cells from functioning as they should, but it is not clear how this leads to white matter damage and cognitive difficulties. Because this mutation is autosomal dominant, only one copy of the altered gene is enough to cause the condition.
The CSF1R gene mutation impacts immune system cells called macrophages and microglia, which normally help protect the brain. In ALSP, this mutation causes:
Misshapen neurons due to bubble-like swellings called spheroids
Loss of myelin as macrophages strip it away from the damaged neurons
Underactive microglia, reducing their normal protective role
This combination of neuron damage, myelin loss, and reduced microglial activity leads to the symptoms seen in ALSP.
While most cases are caused by CSF1R mutations, there are other less common causes:
BANDDOS: Caused by autosomal recessive CSF1R mutations, meaning two altered copies of the gene (one from each parent) are needed. Learn more →
AARS1/AARS2 mutations: Rare changes in these genes can also lead to ALSP. Some AARS2 mutations cause fatal heart disease in infants, while others result in late-onset leukoencephalopathy.
Undetermined mutations: In some patients, ALSP-like symptoms and brain changes appear even when tests for CSF1R and AARS1/AARS2 are negative, suggesting other, yet-undiscovered genetic causes.
Inheritance.
In autosomal dominant conditions, only one copy of the mutated gene is needed to cause the condition. Everyone has two copies of every gene: one from their mother and one from their father.
If a parent has an autosomal dominant condition, they have one normal copy of the gene and one mutated copy. Each child has:
A 50% chance of inheriting the mutated gene (and the condition)
A 50% chance of inheriting the normal gene (and not having the condition)
Because ALSP is often diagnosed later in life, many people have already had children before learning they carry the condition. Currently, there is no newborn genetic screening available for the CSF1R gene mutation that causes ALSP.



Symptoms.
Because ALSP impacts people differently, symptoms can vary widely from person to person, even within the same family. The specific symptoms often depend on the parts of the brain involved.
-

Neurological
DEPRESSION & ANXIETY
MOOD & PERSONALITY CHANGES
MEMORY PROBLEMS
INFLEXIBLE THINKING
LACK OF SELF-CONTROL
POOR JUDGEMENT
LACK OF INTEREST
-

Neurological Cont.
SEIZURES
FATIGUE
INSOMNIA
LOSS OF SPEECH (APHASIA)
VISION CHANGES
IRRITABILITY & CONFUSION
DEMENTIA
-

Muscle & Movement
MUSCLE WEAKNESS & SPASMS
TIGHT MUSCLES
OVERACTIVE REFLEXES
LOSS OF BLADDER/BOWEL CONTROL
MOVEMENT CHANGES
DIFFICULTY WALKING
LACK OF COORDINATION
-

Parkinsonism
RESTING TREMOR
STIFF MUSCLES (RIGIDITY)
SLOW MOVEMENT
SHUFFLING GAIT
LOSS OF FACIAL EXPRESSIONS
DIFFICULTY SITTING/STANDING
DIFFICULTY SWALLOWING

Diagnosis & Treatment.
Diagnosing ALSP can be challenging because symptoms vary greatly from person to person. Doctors may consider additional testing if there is:
A family history of ALSP or related conditions
Early onset of cognitive or movement symptoms (typically before age 60)
Brain imaging showing white matter lesions or other signs of deterioration
ALSP is often misdiagnosed because its symptoms overlap with other conditions, including frontotemporal dementia, multiple sclerosis, Parkinson’s disease, and early-onset Alzheimer’s disease.
A genetic test that looks for changes in the CSF1R or AARS1/AARS2 genes can provide a clear and definitive diagnosis of ALSP.
Currently there are no FDA-approved treatments for ALSP, but there are options to help manage and relieve symptoms. Your healthcare team will partner with you to find the right combination of medications and therapies for your individual needs. Bone marrow stem cell transplants have been performed in some centers and may slow disease progression in ALSP. However, the procedure carries significant risks, including serious infections and immune system complications.



Current Research
Recently Diagnosed?
Navigating a new diagnosis isn’t easy. While it comes with unique challenges, we’re here to remind you that you’re not facing them alone.



